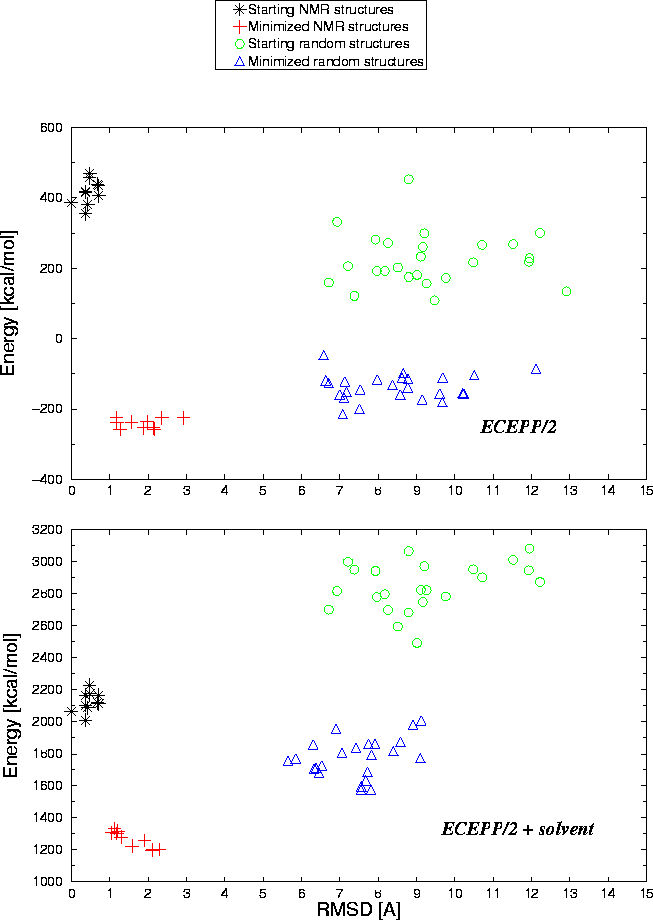
Protein-Solvent Interaction Energy:
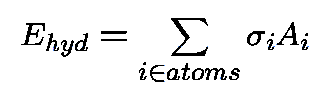
-
 i
i
- : atomic solvation parameter;
- Ai
- : solvent accessible surface area (ASA);
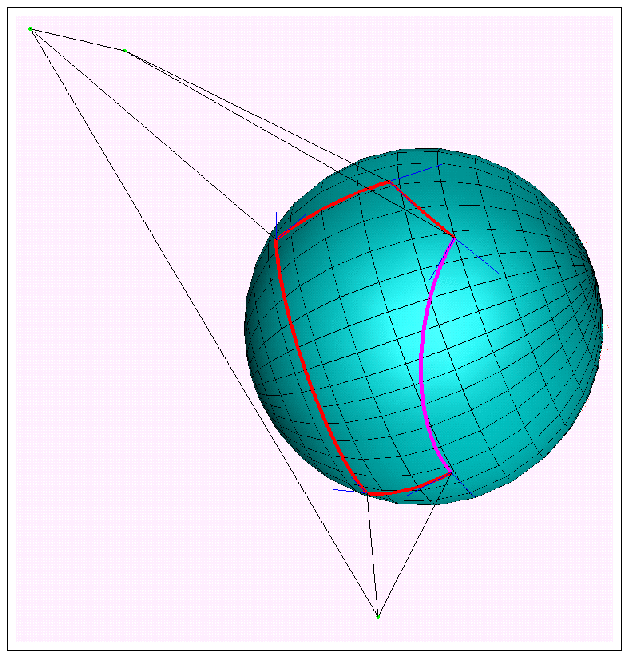
ASA and its derivatives can be calculated analytically from Gauss-Bonnet theorem:
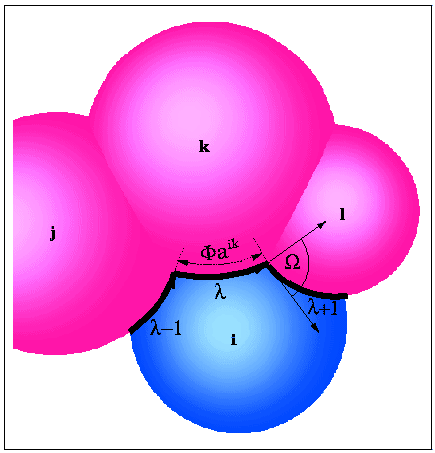
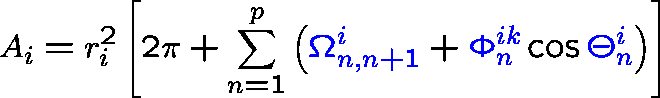
- p
- : number of intersecting arcs defining ASA;
- ri
- : radius of the i-th sphere;
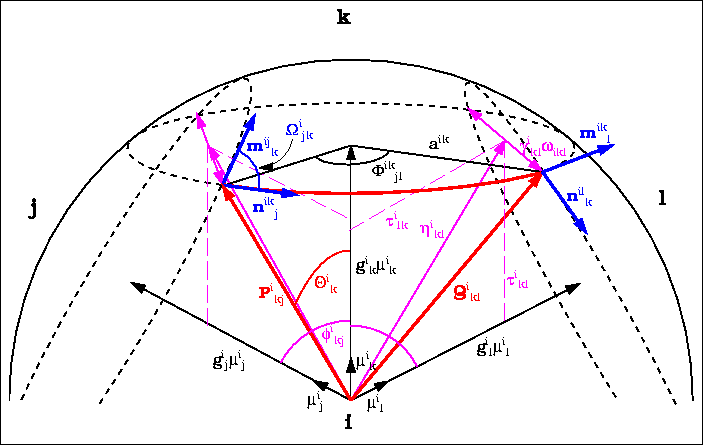
![]() i
n,n+1 :
angle between tangential vectors;
i
n,n+1 :
angle between tangential vectors;
-
 ikn
ikn
- : angle defining arc length;
-
 in
in
- : polar angle of the ik-th intersection circle;
- aik
- : radius of the ik-th intersection circle.